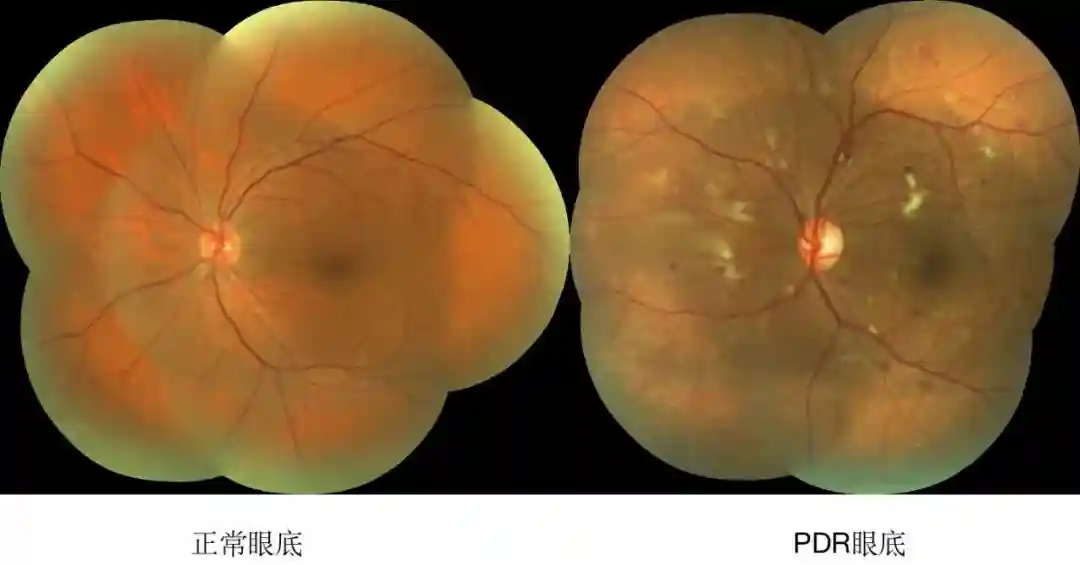
编者按:糖尿病性视网膜病变(DiabeticRetinopathy,DR)是由糖尿病所引起的致盲性眼部并发症,是世界卫生组织(WorldHealthOrganization,WHO)公布的三大致盲性眼病之一,同时也是导致工作人群失明的主要原因[1]。DR可以分为非增殖型(non-proliferativediabeticretinopathy,NPDR)和增殖型(proliferativediabeticretinopathy,PDR)。DR进入增殖期的标志性病理改变是新生血管形成。
近日,一项纳入来自全球35项研究的22896例糖尿病患者的Meta分析显示:在糖尿病患者中,DR的患病率为34.6%,其中PDR患病率为6.96%,糖尿病性黄斑水肿(diabeticmacularedema,DME)患病率为6.81%[2]。我国流行病学调查发现:糖尿病患者中DR的患病率为23%,其中NPDR为19.1%,PDR为2.8%[3]。随着我国糖尿病发病率的逐年增高,DR已成为当前国人主要致盲原因之一[4,5]。CCOS2022会议上,上海交通大学附属第一人民医院刘堃教授带来了其团队在眼底病基础研究的前沿进展。
DR新生血管形成机制的探索
O-GlcNAc糖基化修饰是一种独特的翻译后修饰(post-translationalmodification,PTM)形式,最早于1984年被发现[8]。其独特性体现在,O-GlcNAc糖基化修饰是一种高度动态的、循环的PTM,以UDP-GlcNAc为供体,在N-乙酰氨基葡萄糖转移酶(β-N-acetylglucosaminyltransferase,OGT)的催化作用下,由O-糖苷键连接到蛋白质的丝氨酸/苏氨酸残基上。相反,在N-乙酰氨基葡萄糖苷酶(β-N-acetylglucosaminidase,OGA)的作用下,O-GlcNAc从丝氨酸/苏氨酸残基上水解[9]。外界葡萄糖水平的变化,可以通过己糖胺通路影响UDP-GlcNAc供体,进而影响细胞内蛋白质的O-GlcNAc糖基化修饰。
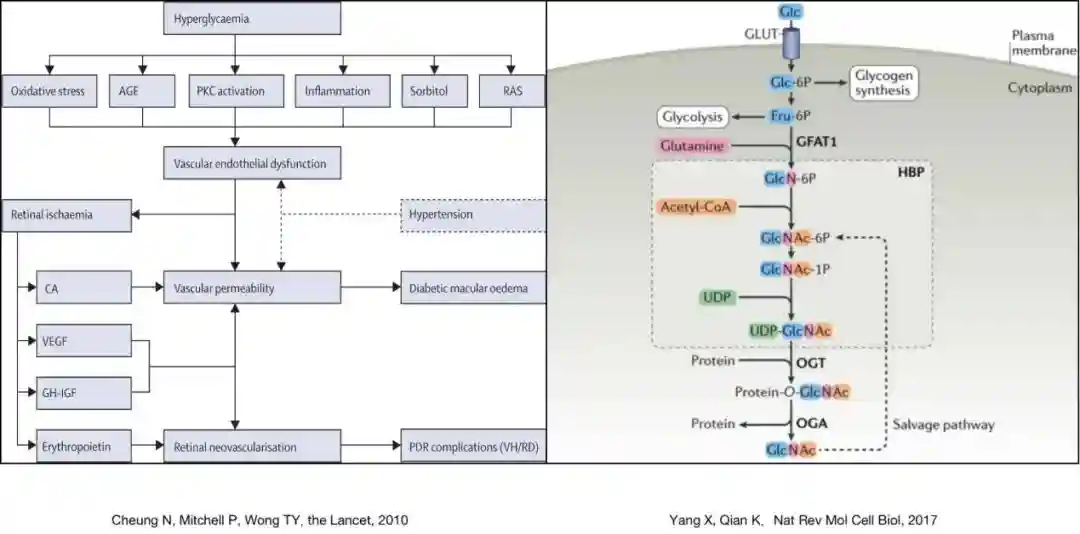
糖尿病最重要的表现是高血糖状态。血糖升高,导致UDP-GlcNAc供体增加,蛋白质的O-GlcNAc糖基化修饰增强。在以往的研究中发现,O-GlcNAc糖基化修饰在糖尿病性心肌病和糖尿病性肾病的发病过程中起到重要作用[10,11],而近期研究指出,O-GlcNAc糖基化修饰在DR进程中也具有类似的作用[12]。在高糖培养的视网膜内皮细胞中,参与糖基化过程的糖基转移酶及其糖基化产物发生改变[13]。在糖尿病鼠视网膜组织中,蛋白质的O-糖基化修饰水平上调[14]。
新生血管形成是DR进入增殖期的标志。视网膜新生血管破裂,引起玻璃体出血,刺激视网膜血管收缩,引起牵拉性视网膜脱离,是造成DR患者视力损害的主要原因[15,16]。新生血管形成是一个动态平衡的过程,受到促血管生成因子如血管内皮生长因子(vascularothelialgrowthfactor,VEGF)以及抗血管生成因子如色素上皮衍生因子(pigmentepitheliumderivedfactor,PEDF)的共同调控[17]。在促血管生成因子作用下,血管内皮细胞增殖、迁移、形成管腔,进而形成新生血管。目前,眼内注射抗VEGF药物治疗DR新生血管取得了较好的治疗效果,但眼内注射抗-VEGF药物有一定的副作用,例如眼内出血、眼内炎、神经网膜损伤、缺血-再灌注损伤等[18-21]。因此,眼科学者们仍然对促进DR新生血管形成的新靶点和新机制进行不懈探索。
以往研究发现,在高糖培养的视网膜细胞中,转录因子特异性蛋白1(transcriptionfactorspecificityprotein1,Sp1)的O-GlcNAc糖基化修饰能促进VEGF的表达[22],VEGF表达上调是DR新生血管产生的直接原因,上述发现表明O-GlcNAc糖基化修饰可能与视网膜新生血管形成存在某种关联。
Runt相关转录因子1(Runt-relatedTranscriptionFactor1,RUNX1)是RUNX转录因子家族的一员,在细胞谱系分化方向的决定、正常造血细胞的形成和干细胞增殖中均发挥重要作用[23]。以往的研究发现,在低氧诱导的视网膜新生血管模型小鼠中,抑制RUNX1能明显抑制模型小鼠视网膜新生血管的形成;在HRMECs中敲除RUNX1,细胞的成管能力下降[24],说明RUNX1有明确的促血管生成作用,但其机制还不明了。
新进展:O-GlcNAc糖基化修饰与RUNX1和DR新生血管之间的关联
基于以上背景,刘教授及其团队研究了O-GlcNAc糖基化修饰与RUNX1和DR新生血管之间的关联:
通过Westernblot实验证实,高葡萄糖条件能够上调视网膜微血管内皮细胞HRMEC的O-糖基化修饰水平,并且在正常糖浓度和高糖情况下添加O-糖基化修饰促进剂,细胞O-糖基化修饰水平随之上调。
分别通过CCK8细胞增殖实验,免疫荧光实验证实,与正常糖浓度相比,高糖培养的视网膜微血管内皮细胞增殖增加,添加O-糖基化修饰促进剂能够进一步促进视网膜微血管内皮细胞增殖,并且通过凋亡指标的Westernblot实验证实,各组之间细胞凋亡没有差别,增殖的增加并不是因为某组凋亡减少才体现出差异。
通过划痕实验,Transwell实验以及成管实验证实,与正常糖浓度相比,高糖能够促进视网膜微血管内皮细胞迁移及成管,并且添加O-糖基化修饰促进剂能够进一步促进视网膜微血管内皮细胞迁移和成管。
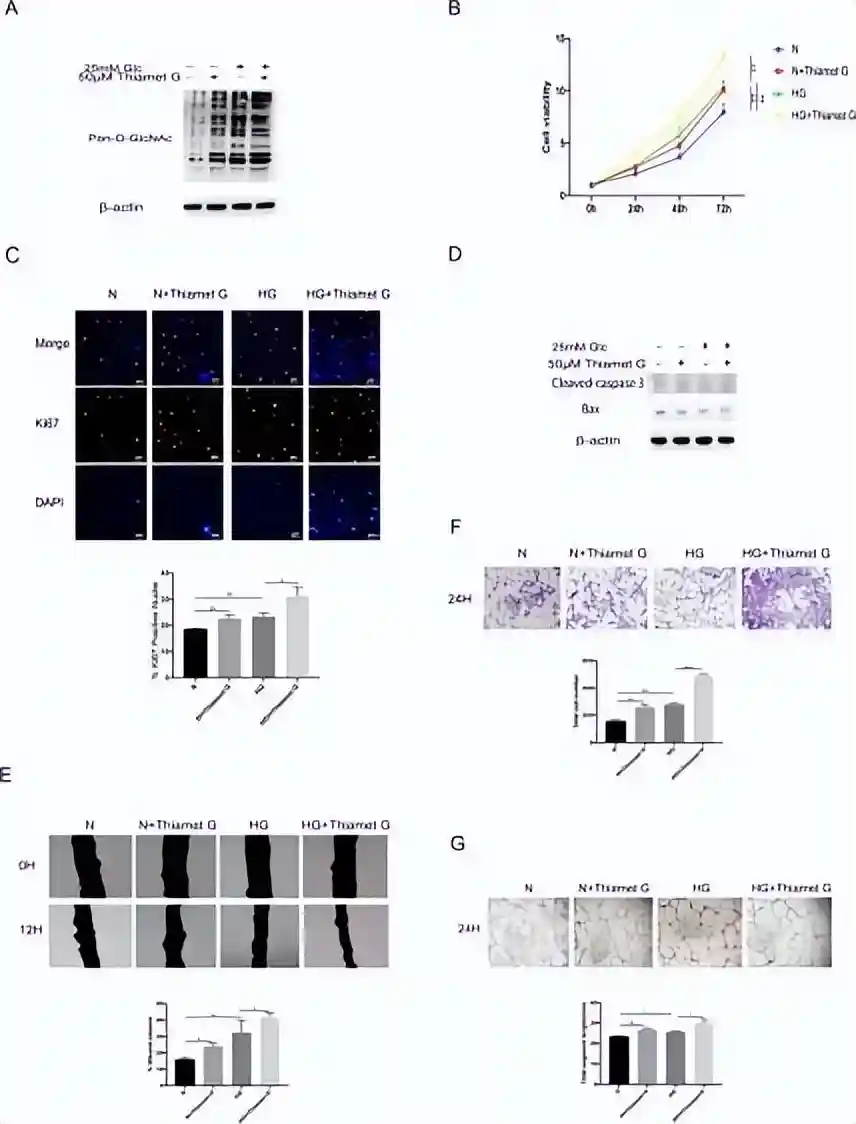
上述实验证实,增加的糖浓度及O-糖基化修饰水平能够促进视网膜微血管内皮细胞的增殖和迁移。
在证实上调葡萄糖浓度及O-糖基化修饰水平能够促进视网膜微血管内皮细胞的增殖和迁移之后,刘教授及其团队又对高糖和O-糖基化修饰的作用进行了反向验证:
分别在正常和高糖条件下添加O-糖基化修饰抑制剂,通过Westernblot实验证实添加的O-糖基化修饰抑制剂能够有效降低细胞的O-糖基化修饰水平。
重复了cck8细胞增殖实验、免疫荧光实验,发现添加O-糖基化修饰抑制剂能够有效抑制视网膜微血管内皮细胞的增殖,并且通过凋亡指标的Westernblot实验证实,各组之间细胞凋亡没有差别,增殖的减少并不是因为某组细胞凋亡增加才体现出差异。
进行了划痕实验、Transwell实验以及成管实验,证实下调O-糖基化修饰水平后,细胞的迁移和成管能力随之下调。
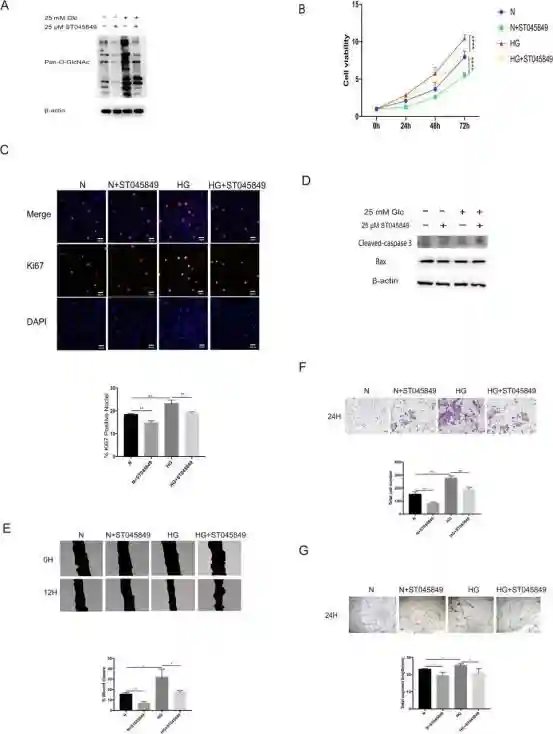
以上实验证实,随着O-糖基化修饰水平降低,视网膜微血管内皮细胞的增殖和迁移能力也下降。
证实了O-糖基化修饰的作用以后,刘教授及其团队继续探索RUNX1在这个过程中的作用:
通过两条不同的siRNA序列对细胞中的RUNX1进行敲减,通过Westernblot实验证明敲减有效。
分别在正常糖浓度、高糖浓度、添加和不添加O-糖基化修饰促进剂、敲减和不敲减RUNX1的情况下进行CCK8和免疫荧光实验,证实在敲减RUNX1之后,高糖和高O-糖基化修饰水平对视网膜微血管内皮细胞增殖的促进作用都减弱了。
分别在正常糖浓度、高糖浓度、添加和不添加O-糖基化修饰促进剂、敲减和不敲减RUNX1的情况下进行划痕实验、Transwell实验以及成管实验,发现敲减RUNX1之后,高糖和高O-糖基化修饰水平对视网膜微血管内皮细胞迁移和成管的促进作用也减弱了。
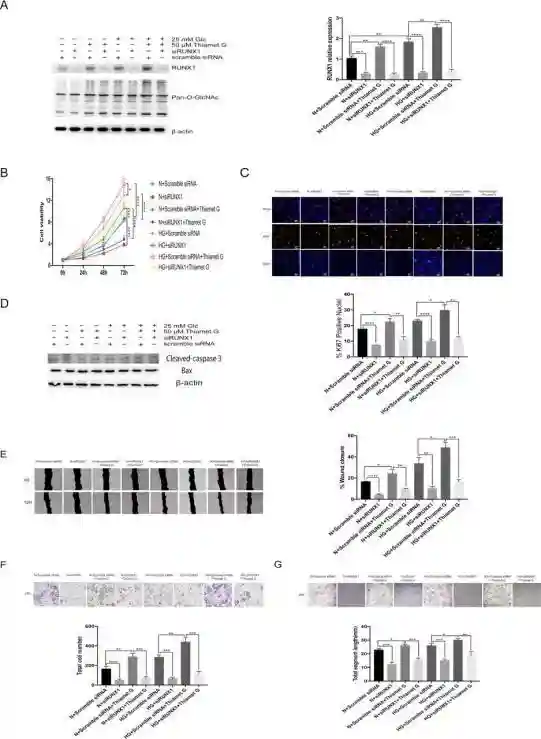
通过上述实验,证实高糖和高O-糖基化修饰水平对视网膜微血管内皮细胞增殖和迁移的促进作用必须有RUNX1存在。
刘教授及其团队进而探索了其可能机制:
用STZ诱导糖尿病大鼠模型,升高的血糖、降低的体重和紊乱的视网膜结构证实造模成功。取视网膜进行了免疫共沉淀实验,发现糖尿病大鼠视网膜组织中,RUNX1的O-糖基化修饰增强。对细胞也进行了免疫共沉淀实验,发现在高糖培养的细胞中,RUNX1的O-糖基化修饰增强,而添加O-糖基化修饰促进剂后,RUNX1的O-糖基化修饰进一步增强,因此推测O-糖基化通过修饰RUNX1影响视网膜血管内皮细胞的增殖和迁移,进而影响新生血管形成。
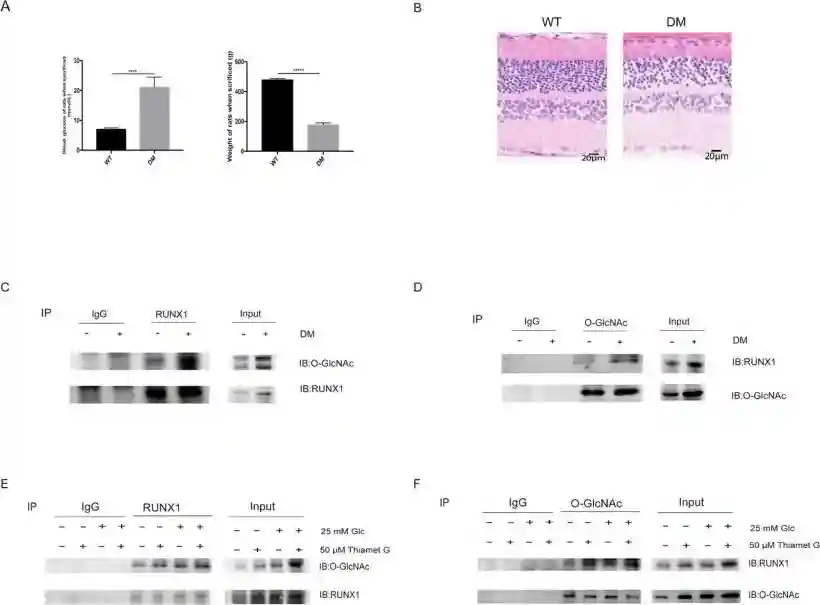
通过上述实验,证实了O-糖基化修饰通过RUNX1对DR新生血管的促进作用。
小结:未来,可通过对修饰位点的测序和突变进行更深入的机制探究,期待有更多新的发现,还DR患者光明视界。
参考文献
[1]KleinBE,Overviewofepidemiologicstudiesofdiabeticretinopathy[J].
OphthalmicEpidemiology2007,(14):179–183.
[2]YauJW,RogersSL,KawasakiR,
ofdiabeticretinopathy[J].DiabetesCare,2012,35(3):556-564.
[3]LiuL,WuX,LiuL,
:ameta-analysis[J].PLoSOne,2012,7(9):e45264.
[4]白雪林,徐娴,陆敏等.上海市新泾社区2型糖尿病患者盲和中重度视觉损伤的流行病学
调查.中华眼科杂志,2016,52(11):825-830。
[5]管怀进.我国防盲与眼科流行病学研究的现状及发展[J].中华眼科杂
志,2010,46(10):938-943.
[6]JuanLi,YongHuaHu,Susceptibilitygenesfordiabeticretinopathy[J].
InternationalJournalofophthalmology2(2008)234-239.
[7],DiabeticRetinopathy[J].TheNewEnglandJournalofMedicine
350(2004)48-58.
[8]TorresCR,
linkedGlcNAc[J].(1984)3308–3317.
[9]章圆,刘堃,陈凤娥,0位N一乙酰葡萄糖胺糖基化修饰与糖尿病视网膜病变的相关性研
究现状[J].中华眼底病杂志,2017,33(
7)318-321.
[10]YokoeS,AsahiM,TakedaT,
byO-GlcNAcylation:implicationsfordiabeticcardiomyopathy[J].Glycobiology.
2010,20(10):1217-1226.
[11]ParkMJ,KimDI,LimSK,
carbohydrateresponseelement-bindingprotein(ChREBP)mediatesmesangialcell
lipogenesisandfibrosis:thepossibleroleinthedevelopmentofdiabetic
nephropathy[J].,289(19):13519-13530.
[12]PetersonSB,:AroleforO-GlcNAcylationindiabetic
complications[J].,51(3):150-161.
[13]LiuK,LiuH,ZhangZ,
inducedupregulationofintercellularadhesionmolecule-1onbovineretinal
othelialcells[J].,94(4):353-357.
[14]GurelZ,ZaroBW,PrattMR,
targetsinmouseretinalpericytes:implicationofp53inpathogenesisofdiabetic
retinopathy[J].,9(5):e95561.
[15]AielloLP,AveryRL,ArriggPG,
inocularfluidofpatientswithdiabeticretinopathyandotherretinal
disorders[J].,331(22):1480-1487.
[16]SchroderS,PalinskiW,
granulocytes,capillarynonperfusion,andneovascularizationindiabetic
retinopathy[J].,139(1):81-100.
[17]FolkmanJ,[J].SeminarsinCancerBiology.
1992,(3):89–96.
[18]HsuYJ,HsiehYT,YehPT,HuangJY,YangCM,CombinedTractionaland
RhegmatogenousRetinalDetachmentinProliferativeDiabeticRetinopathyinthe
Anti-VEGFEra[J].:917375.
[19]KuiperEJ,
diabeticretinopathy[J].,3(7):e2675.
[20]Saint-GeniezM,:
evidenceforasurvivalroleonmullercellsandphotoreceptors[J].PLoSOne2008,
3(11):e3554.
[21]SangDND'AmorePAIsblockadeofvascularothelialgrowthfactor
beneficialforalltypesofdiabeticretinopathy?[J].,
51(9):1570-1573.
[22]SherZamanSafi,RajesQvist,SelvaKumar,
DiabeticRetinopathy,GeneralPreventiveStrategies,andNovelTherapeutic
Targets[J].:801269
[23]LeeSH,
Angiogenesis[J].AdvancesinExperimentalMedicineandBiology,2017,(962):449-
469.
[24]LamJD,OhDJ,WongLL,etal,IdentificationofRUNX1asamediatorof
aberrantretinalangiogenesis,,(66):1950-1956.



